
Interprofessional Management of Hypophosphatasia
The oral complications of this rare condition underscore the need for multidisciplinary collaboration among health care teams.
The oral complications of this rare condition underscore the need for multidisciplinary collaboration among health care teams
PURCHASE COURSE
This course was published in the April 2017 issue and expires April 2020. The authors have no commercial conflicts of interest to disclose. This 2 credit hour self-study activity is electronically mediated.
OBJECTIVES
After reading this course, the participant should be able to:
- Discuss the prevalence of hypophosphatasia (HPP) and the U.S. populations most commonly affected.
- List common histories, symptoms and radiographic findings in patients with HPP.
- Describe the various forms of HPP.
- Explain clinical approaches to managing this patient population.
Collaboration among the extended health care team is crucial for patients’ overall health and well-being, and this cooperative perspective is often best seen when an oral complication is indicative of other systemic conditions. In light of the growing need for interprofessional collaboration — and due to the possibility of favorable outcomes even with complex diseases — this paper will explore a relatively rare metabolic condition, hypophosphatasia (HPP), and present a corresponding case report.
This inherited disorder is characterized by a spectrum of defects caused by mutations of the alkaline phosphatase gene (ALPL). The gene codes for an enzyme called tissue-nonspecific alkaline phosphatase (TNSALP), which is abundant in the skeleton, liver, kidneys and developing dentitions.1,2 These mutations, often missense mutations (i.e., a single nucleotide change resulting in a different amino acid), result in low TNSALP activity. With such low TNSALP biochemical defects, bone and tooth dysplasia are common clinical findings.3 Reports of HPP prevalence vary; estimates range from one in 2500 individuals to one in 100,000. Among the U.S. population, it is most commonly found in Caucasians.3
This condition’s high variability in clinical presentation is attributed to allelic heterogeneity in ALPL. Furthermore, severe forms of HPP are inherited in an autosomal recessive pattern (two copies of the gene are altered), while less severe forms have either autosomal recessive or autosomal dominant (only one copy of the gene is altered) inheritance patterns.4 Because of this inheritance transmission, and the known 300-plus ALPL mutations, individuals with HPP have significant ranges in clinical expression — the exception being premature tooth loss, which is a common presenting symptom. To that end, oral health care providers serve a unique role in early recognition of this complex and debilitating disease.
Until recently, there was no treatment for HPP, but with the advent of a new enzyme replacement therapy, the dental professional’s role in identifying these cases for early referral, diagnosis and treatment is more important than ever.
PATHOPHYSIOLOGY
In healthy infants and children, TNSALP is abundant in bone, while in adults it is found in the kidneys, liver and bone.3 Although the effects of deficient or low levels of TNSALP are known in mineralized tissues (including teeth), its effect on the kidneys and liver is not understood.3 Biochemically, TNSALP cleaves phosphorylated substrates, such as inorganic pyrophosphate (PPi), pyridoxal-5-phosphate (PLP), and phosphoethanolamine (PEA), and is expressed in the liver, kidneys, brain, teeth and bone.
The TNSALP enzyme initiates the process of bone matrix mineralization through the dephosphorylation of PPi into Pi. Deficiency of TNSALP facilitates the extracellular accumulation of PPi, leading to defects in bone and teeth mineralization.5 Specifically, PPi acts by blocking the synthesis of hydroxyapatite crystals during bone mineralization.
The PLP substrate is the phosphorylated form of vitamin B6, and deficiency in the TNSALP enzyme causes a decrease in the dephosphorylation of PLP to pyridoxal — a necessary step for pyridoxine to be transported across the blood-brain barrier. Increases in PLP levels suggest impaired transport of vitamin B6 to the brain, and, in severe forms of HPP, can lead to seizures.4
DIAGNOSIS
Biochemical Markers and ALPL Molecular Testing — Obtaining laboratory values is among the first steps in a definitive HPP diagnosis. Affected individuals have consistently low serum alkaline phosphatase (ALP) activity for their age and sex. A single low value is not sufficient to establish a diagnosis, as it can present in other conditions, such as celiac disease, severe anemia, hypothyroidism, hypoparathyroidism, Wilson’s disease, hypomagnesemia and malnutrition, as well as with vitamin C, folate or vitamin B12 deficiencies. Use of medications, including bisphosphonates, glucocorticoids, estrogens, omeprazole, clofibrate and high doses of vitamin D, have also been associated with low ALP.
Patients with HPP typically have elevated levels of phosphorylated substrates, such as PLP and urinary PEA. Elevated PEA levels are not pathognomonic (as can be seen in other metabolic bone diseases) and may be normal for some individuals with HPP. Transient abnormalities include hypercalcemia, hyperphosphatemia, low parathyroid hormone levels and hypercalciuria, leading to nephrocalcinosis. Upon finding suspicious abnormal laboratory values, sequencing of the ALPL gene and assessment for mutations will help confirm the diagnosis. There is strong correlation between molecular testing and TNSALP enzymatic activity.6
Radiographic Features — In utero-affected fetuses have decreased bone mineralization, short and/or bowed long bones, and spur-like projections of the long bones. Stillborn infants typically have irregular, short and deep cup-shaped metaphyses, hypoplastic or absent bones, and thin diaphysis. Characteristic radiographic findings in infants and children with HPP include rickets-like changes, decreased bone mineralization, dolichocephalic facial features, undermineralized skull, craniosynostosis, enlarged and irregular metaphyses, and metaphyseal radiolucencies. The diaphyses may show fractures or zones of osteosclerosis.
Adults may have numerous stress fractures of the metatarsals, femoral pseudofractures, delayed healing of fractures, focal osteolysis, chondrocalcinosis, leg bowing and periarticular calcifications. Bone scintigraphy may help to screen for stress fractures. Decreased mineralization can be quantified by densitometry.
CLINICAL PRESENTATION
Although HPP’s clinical manifestations are extremely variable, they frequently involve a defect in the mineralization of the bone and/or teeth. This disease is characterized by the age at which clinical symptoms develop into six forms: perinatal (lethal), perinatal benign, infantile, childhood, adult and odontohypophosphatasia.4,7 This classification does not, however, reflect that these forms are part of phenotypic continuum of HPP, and that the disease can evolve as an individual ages. Children with HPP may have no symptoms other than tooth loss, but may present later in life with unexplained fractures, bone pain and osteopenia.8
Perinatal HPP is the most severe form of HPP. Clinical consequences are established in utero and the condition is lethal soon after birth. Among other features, these patients exhibit generalized skeletal hypomineralization, seizures, respiratory complications and intracranial bleeding. Perinatal benign HPP includes individuals who show spontaneous improvement during the third trimester of pregnancy, or soon after birth. Careful evaluation, including radiographic, biochemical and molecular testing, is needed to distinguish between perinatal benign HPP and perinatal lethal HPP.
There have been reports of patients with HPP whose bone abnormalities improve postnatally. It has also been shown to be present in infancy or later in childhood with failure to thrive, delayed motor milestones, bone pain, leg bowing, joint enlargement (similar to that of rickets), craniosynostosis, impaired growth, impaired mineralization and fractures. Craniosynostosis may be present at birth. Motor delays and waddling gait have been attributed to muscle hypotonia. In severe cases, impaired hearing can develop over time. Fatigue, absence of a pubertal growth spurt, or bone pain and fractures are presenting symptoms of HPP in adolescents and adults.
Among the adult population, HPP can manifest in middle age. A history of premature loss of deciduous teeth, with loss of adult dentition, is common.9 Of note, some patients may have a history of premature tooth loss, followed by uneventful childhoods until they develop symptoms as adults. This underscores the disease’s evolution and progression over the years, and highlights the need for life surveillance of children with isolated tooth loss and HPP. Adult patients often report bone fractures that have failed to heal, bone pain and muscle weakness, and can present with ligament ossification.
ORAL FEATURES
The hallmark of the disease is increased tooth mobility and premature, painless loss of the primary teeth (with intact roots) that is not due to trauma. This is due, in part, to cementum that ranges from thin to completely unformed (i.e., aplastic).10 The incisors are often the first to be affected and are typically lost by 2 years of age. Depending on the severity of disease, most individuals lose more teeth at a younger age, starting with the lower and upper anterior teeth. Oral manifestations are the only symptoms in some patients, and are characterized by dento-osseous changes depicted by alveolar bone loss, as well as dentin and cementum dysplasia.10 Other manifestations include delayed tooth eruption, enamel hypoplasia, and enlarged root canals and pulp chambers.11 Defects in cementum and reduced marginal alveolar bone are thought to contribute to premature tooth exfoliation. Thus, HPP should be considered in any patient with a history of early, unexplained loss of teeth or abnormally loose teeth.
MEDICAL MANAGEMENT AND TREATMENT
Pharmacologic Management — Nonsteroidal anti-inflammatory drugs are typically used to treat pain and inflammatory symptoms, even during childhood. Clinicians should monitor vitamin D, calcium and phosphate serum levels, and high levels should be avoided, as this can lead to hypercalcemia and nephrocalcinosis. The use of bisphosphonates is contraindicated, however, and likely to worsen clinical outcomes.12
Enzyme Replacement — Enzyme replacement is a promising new therapy available to individuals with HPP that was recently approved by the U.S. Food and Drug Administration. It consists of engineered asfotase alfa, a recombinant ALP fusion protein, and is available at a dose of 2 mg/kg three times a week, or 1 mg/kg/day six times a week in children. Preclinical studies of ALP-deficient mice and clinical studies in infants and children have shown notable improvement in bone mineralization, muscle function, respiratory function and height z-scores.13,14 Local site reactions, such as erythema, pruritus and pain, are the most frequent side effects.
DENTAL MANAGEMENT
Currently, there are no standardized guidelines to address dental complications in patients with HPP. Like other patients with special health care needs, maintaining optimal oral hygiene is critical because biofilm accumulation may contribute to the loss of already fragile dentition.7,15 Oral hygiene instruction and appropriate dietary counseling are the best preventive measures for slowing the process of premature tooth loss. Few case reports in the literature characterize treatment of partial anodontia in individuals with HPP. Prosthodontic rehabilitation has been limited to young adults and adults by using partial dentures, overdentures and dental implants.11,16 Although the long-term prognosis of such treatment is not well understood, these options address esthetics, functional characteristics of occlusion and speech, and mental depression. Periodic examination to assess mobility of remaining abutment teeth is important because partial or overdentures could potentially compromise remaining dentition.
Because enzyme replacement therapy (using alfotase alfa as a treatment option) is relatively new, there are no reports examining its potential effect on dental outcomes. Little is known about the potential prevention of premature tooth loss via effects on cementum development and reduced alveolar bone loss for patients who are treated with enzyme replacement therapy. Future research is needed to study these effects.
CASE REPORT
A 19-month-old girl was referred to a University of Minnesota pediatric dental clinic for consultation due to the unexplained, nontraumatic, premature loss of the patient’s lower central incisors. At the initial consult, the patient presented with an unremarkable medical history and was in good general health. She was growing along the 14th percentile for length and 34th percentile for weight. She lost her first tooth at 10 months of age, and a second tooth at 12 months. At 15 months, she was found to have four loose teeth. Although there was no family history of skeletal abnormalities, her maternal grandfather and his mother started losing teeth in their 50s. The patient’s 6-year-old brother had no history of premature tooth loss.
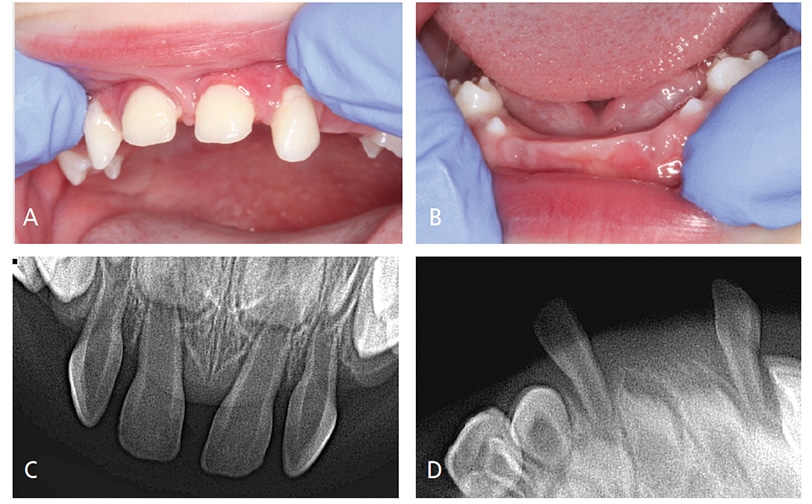
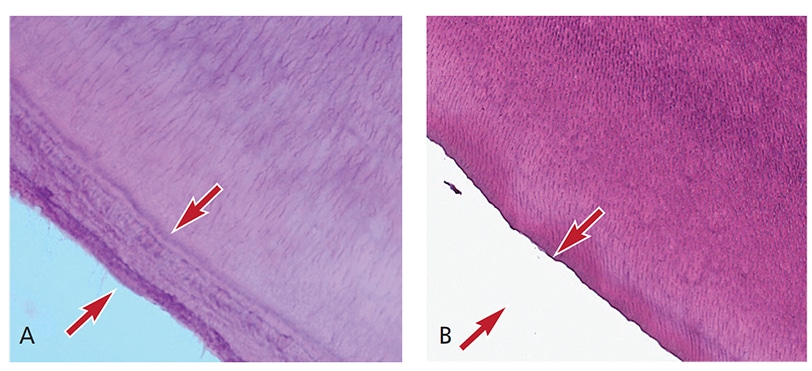
Oral Evaluation — Initial interpretation and assessment by a general dentist attributed the premature loss of her first primary incisor to unwitnessed orofacial trauma. After the loss of her second primary incisor and presence of four loose maxillary incisors, the patient was evaluated by a pediatric dentist, who suspected HPP and referred her to a medical provider. At the first medical consultation, the potential diagnosis of HPP was dismissed due to low, yet within normal, levels of alkaline phosphatase. Subsequent evaluations of alkaline phosphatase levels were also low at 18 months, and again two weeks later. She subsequently exfoliated a third primary tooth — a lower lateral incisor. At that time, the patient was referred to the university dental clinic, where a comprehensive evaluation, including radiographic examination, was performed (Figures 1A through 1D). The third exfoliated tooth was submitted to the University of Minnesota Oral Pathology Laboratories for histologic assessment. The lab confirmed a lack of cementum (Figures 2A and 2B), which is consistent with a potential diagnosis of HPP.
The patient was subsequently referred to a pediatric endocrinologist for further evaluation.
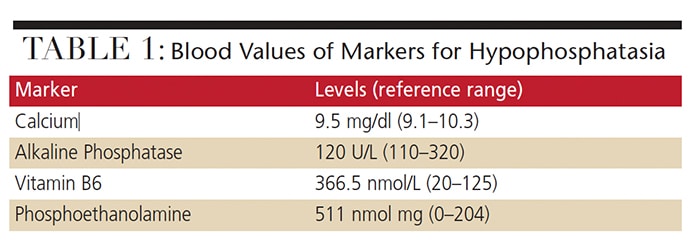
Biochemical and Radiographic Evaluation — The patient’s urinary PEA and PLP levels were elevated (Table 1). Her vitamin D, intact parathyroid hormone, calcium and phosphorus levels were all within normal range, however. In addition, her bone alkaline phosphatase level was low (30.1 ug/L, compared to a normal range of 33.4 to 145.3 ug/L). Radiographic imaging showed no skeletal abnormalities.
Genetic Analysis — Gene sequencing found a heterozygous mutation c.1259G>C (pGly420Ala) in the ALPL gene. Given the lack of a second pathogenic mutation, clinical findings and published evidence suggest this mutation acts in a dominant negative manner, and indicate a diagnosis of autosomal dominant HPP. This mutation has been described in patients with mild HPP who are heterozygous only for this mutation. Furthermore, ALPL gene sequencing of the child’s mother found the same heterozygous mutation as her daughter’s. The mother has a history of pain in her metatarsal and femoral bones.
Dental Management — As of this report, the patient has been followed without incident every six months. Dut to her age (35 months), oral/dental management has focused on preventive measures, including oral hygiene assessment and dietary counseling. A plan is not yet in place for replacement of premature exfoliated teeth. Anticipatory guidance has been explained to the parents, including the potential effects of HPP on the patient’s permanent dentition.
CONCLUSION
A number of systemic disorders are associated with the premature loss of primary dentition, including Papillion-Lefevre syndrome, Chediak-Higashi syndrome, neutropenia, leukemia, Langerhans cell histiocytosis and HPP. In the absence of trauma, primary tooth exfoliation (with intact roots) occurring prior to the expected exfoliation period should raise suspicion among the extended health care team of a systemic condition. Once diagnosed, multidisciplinary collaboration with specialized medical and dental practitioners is important to proper management of HPP. Under such collaborations, appropriate recommendations, anticipatory guidance, follow-up and management will help ensure the best possible care and clinical outcomes.
REFERENCES
- Millán JL. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley-VCH Verlag GmbH; 2006.
- Thakker RV, Whyte MP, Eisman JA, Igarashi T. Genetics of Bone Biology and Skeletal Disease. San Diego, Ca: Academic Press; 2013:3–146.
- Whyte MP. Hypophosphatasia — aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12:233–246.
- Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007;2:40.
- Millán JL, Plotkin H. Hypophosphatasia — pathophysiology and treatment. Actual Osteol. 2012;8:164–182.
- Mornet E. Molecular genetics of hypophosphatasia and phenotype-genotype correlations. Subcell Biochem. 2015;76:25–43.
- Olsson A, Matsson L, Blomquist HK, Larsson A, Sjödin B. Hypophosphatasia affecting the permanent dentition. J Oral Pathol Med. 1996;25:343–347.
- Linglart A, Biosse-Duplan M. Hypophosphatasia. Curr Osteoporos Rep. 2016;14:95–105.
- Berkseth KE, Tebben PJ, Drake MT, Hefferan TE, Jewison DE, Wermers RA. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone. 2013;54:21–27.
- Lundgren T, Westphal O, Bolme P, Modeer T, Noren JG. Retrospective study of children with hypophosphatasia with reference to dental changes. Eur J Oral Sci. 1991;99:357–364.
- Grewal PS, Gupta KP. Prosthetic rehabilitation of a young patient with hypophosphatasia: a review and case report. Contemp Clin Dent. 2012;3:74–77.
- Sutton RA, Mumm S, Coburn SP, Ericson KL, Whyte MP. “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Miner Res. 2012;27:987–994.
- Whyte MP, Greenberg CR, Salman NJ, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366:904–913.
- Millán JL, Narisawa S, Lemire I, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23:777–787.
- el-Labban NG, Lee KW, Rule D. Permanent teeth in hypophosphatasia: light and electron microscopic study. J Oral Pathol Med. 1991;20:352–360.
- Lynch CD, Ziada HM, Buckley LA, O’Sullivan VR, Aherne T, Aherne S. Prosthodontic rehabilitation of hypophosphatasia using dental implants: a review of the literature and two case reports. J Oral Rehabil. 2009;36:462–468.
Featured photo by Yarinca / istock / Getty Images Plus
From Decisions in Dentistry. April 2017;3(4):50–53.
![The controversy around adding fluoride to drinking water was recently highlighted by an article in the Wall Street Journal, which addressed its possible negative neurological effects on children.[1] The article cited recent studies on what levels of fluoride are needed to protect teeth without risking possible cognitive harm to prenatal children and infants. Link in bio.
[1] Data on link.
---
#dentistry #dentist #dental #smile #dentista #teeth #cosmeticdentistry #dentistryworld #dentalphotography #odonto #tooth #dentistrylife #orthodontics #dentalcare #dentalhygienist #dentalimplants #oralhealth #veneers #dentalstudent #dentalassistant #dentalclinic #dentistlife #dentalhygiene #teethwhitening #oralsurgery #dds #endodontics #continuingeducation #education](http://decisionsindentistry.com/wp-content/plugins/instagram-feed/img/placeholder.png)
