
Managing Caries Risk in Patients With Cystic Fibrosis
Assessing specific risk factors will help clinicians support the oral health of this patient population.
Assessing specific risk factors will help clinicians support the oral health of this patient population
PURCHASE COURSE
This course was published in the August 2016 issue and expires 08/31/19. The author has no commercial conflicts of interest to disclose. This 2 credit hour self-study activity is electronically mediated.
OBJECTIVES
After reading this course, the participant should be able to:
- Discuss the prevalence and presentation of cystic fibrosis (CF).
- Identify this population’s risk factors for dental caries.
- Explain the appropriate clinical considerations when treating
patients with CF.
Cystic fibrosis (CF) is the most common lethal genetic disease in Caucasians. Of the 70,000 individuals diagnosed with CF worldwide, about 30,000 live in the United States.1 Approximately 1000 individuals are diagnosed with CF in the U.S. each year. Because CF is caused by an autosomal gene mutation, males and females are equally affected, although the disease incidence varies by race. The vast majority of individuals with CF are Caucasian, with a prevalence of one in 3200. It occurs less often in other races, with a reported prevalence of one in 7000 among Hispanics, one in 15,000 among African Americans, and one in 31,000 among Asian Americans.2,3
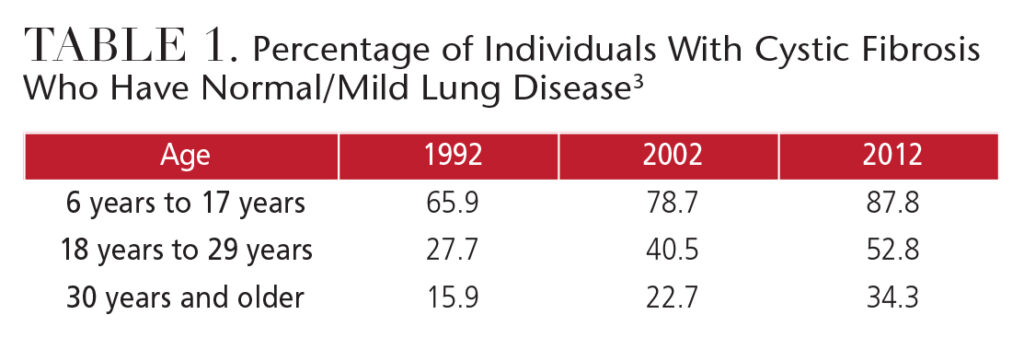
This disease is caused by an autosomal recessive mutation to the cystic fibrosis transmembrane regulator (CFTR) gene. The CFTR gene defect disrupts chloride and sodium ion movement across cell membranes, which results in thick mucus obstructions. The most damaging obstruction occurs in the lungs (Table 1), leading to adverse health effects, including infections that can progress to irreversible lung disease and, ultimately, death.3,4 Progression of CF with mild to moderate lung disease to CF with severe lung disease occurs over periods of stable health, with marked, intermittent periods of acute disease.
Historically, CF diagnosis was based on clinical signs, including frequent lung infections, abnormally salty skin, and failure to thrive. The presence of meconium ileus (complete bowel obstruction) shortly after birth once served as a screening tool.5 Today, nearly 60% of individuals with CF are diagnosed by newborn screening panels that are mandated in all 50 states.3 The diagnosis is confirmed by a skin sweat test and genetic testing.
 Although CF affects multiple organ systems, including the sinuses, lungs, gastrointestinal tract and pancreas, the most damaging effects are on the lungs. As thickened secretions build up in the lungs, pathogenic bacteria colonize the mucus, which, in turn, stimulates the production of additional mucus. This can lead to lung collapse, permanent scarring of lung tissue and death.6 There is no cure for CF. Aggressive treatment protocols consist of airway clearance techniques, systemic antibiotics and nutritional approaches — all of which have markedly improved prognosis and survival rates. Managing CF requires a coordinated, team-based approach. Thus, most individuals with CF receive medical care within certified CF centers, and meet regularly with members of a multidisciplinary team for both acute and routine care. These teams include physicians, pulmonologists, nurses, physical therapists, nutritionists, social workers, genetic counselors, pharmacists, psychologists and infectious disease specialists.
Although CF affects multiple organ systems, including the sinuses, lungs, gastrointestinal tract and pancreas, the most damaging effects are on the lungs. As thickened secretions build up in the lungs, pathogenic bacteria colonize the mucus, which, in turn, stimulates the production of additional mucus. This can lead to lung collapse, permanent scarring of lung tissue and death.6 There is no cure for CF. Aggressive treatment protocols consist of airway clearance techniques, systemic antibiotics and nutritional approaches — all of which have markedly improved prognosis and survival rates. Managing CF requires a coordinated, team-based approach. Thus, most individuals with CF receive medical care within certified CF centers, and meet regularly with members of a multidisciplinary team for both acute and routine care. These teams include physicians, pulmonologists, nurses, physical therapists, nutritionists, social workers, genetic counselors, pharmacists, psychologists and infectious disease specialists.
CARIES RISK AND CYSTIC FIBROSIS
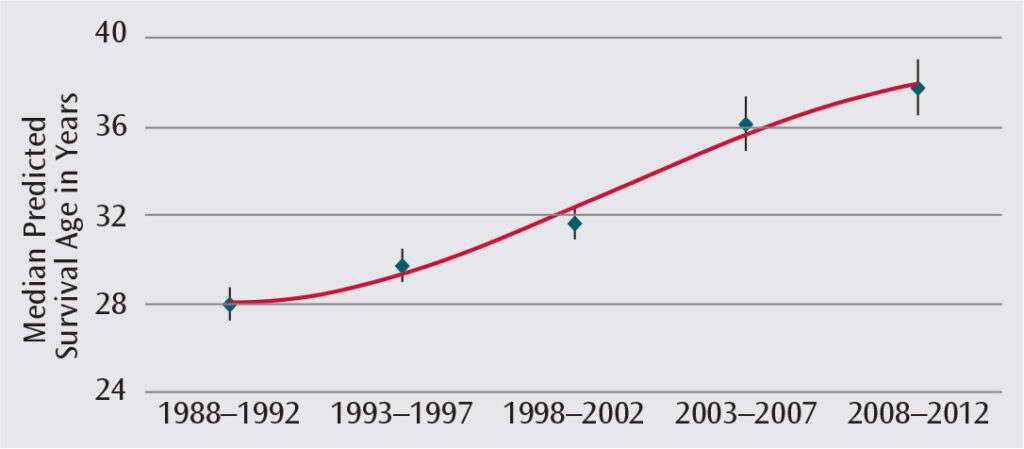
The life expectancy of individuals with CF has increased significantly in recent decades (Figure 1).3 In the 1950s, most patients diagnosed with CF did not survive beyond elementary school. In 1992, the predicted mean survival was 29.4 years. This increased to 41.1 years in 2012. Currently, many individuals with CF live into their 50s and 60s.3 About half of individuals with CF in the U.S. are younger than 18.
Historically, dental caries may not have been a high priority health problem among those with CF because of shortened life expectancy. Recent improvements in the management of CF and the subsequent increase in life expectancy, however, mean that caries prevention should be considered an integral part of comprehensive infection prevention plans. This is important in ensuring quality of life and optimal overall health.
In order to help individuals with CF prevent caries, oral health professionals need to understand the specific risk factors that affect pediatric patients with CF — and how to manage caries risk in children and adolescents with CF.
Often present on the maxillary incisors, enamel defects rank among the earliest reported dental anomalies described in individuals with CF. The prevalence of enamel defects ranges from 28.4% to 56.0%.7–11 Mouse models suggest that enamel defects are likely caused by the abnormal expression of the CFTR gene in cells that produce enamel.12 The CFTR gene appears to play a role in enamel formation; specifically, it is turned on as ameloblasts mature and is critical for completion of the mineralization process.13
Gastrointestinal complications are common in patients with CF. They frequently begin in the neonatal period and may progress to digestion problems and gastroesophageal reflux disease (GERD).14 About 55% to 76% of individuals with CF are diagnosed with GERD, which can exacerbate respiratory disease.15,16 Backflow of stomach contents into the esophagus irritates tissues in the larynx, trachea and bronchi, leading to chronic coughing. In addition, GERD can force stomach acid into the oral cavity, which, depending on frequency and severity, can erode the dentition.17 Erosion from GERD decreases the integrity of the mineral component of the enamel matrix, predisposing affected teeth to caries.18 Studies comparing children with GERD and their healthy siblings show significantly higher caries levels in those with GERD.19 These effects are likely to be more pronounced in children and adolescents with CF because of enamel defects secondary to CFTR defects.
Although saliva helps protect teeth by buffering against the harmful effects of dietary sugars, the CFTR gene also affects the salivary glands of individuals with CF. Clinical manifestations include salivary gland hypertrophy8 and structural alterations, especially in the mucus-producing sublingual gland.6 A study of young children with CF reported significantly higher salivary pH (i.e., more basic saliva) and increased buffering capacity in saliva expressed during mealtimes,20 but studies of resting salivary pH and buffering capacity are not available. Saliva from the submandibular gland of mice without the CFTR gene has a lower pH, however, suggesting that the defective CFTR gene may disrupt secretion of bicarbonate — an important buffering ion in saliva — and lead to a greater number of, and more severe, caries lesions.21 More studies are needed to fully assess the CFTR gene’s effect on salivary pH.
CALORIC INTAKE
Absorption of nutrients is a challenge for patients with CF because of the thickened mucous lining of the intestinal tract, combined with CFTR gene-mediated pancreatic insufficiency. Nutritionists play a key role in counseling parents/caregivers of children with CF. Children with CF are advised to increase calorie intake to ensure proper weight gain.22,23 Up to 200% of the daily recommended calories should come from a balanced diet high in protein and fats. Anecdotally, a large number of children with CF may rely on refined carbohydrates to meet calorie requirements. The combination of cariogenic food consumption and increased eating frequency may further raise the caries risk for individuals with CF.24
To maintain respiratory health, individuals with CF routinely take a number of medications. Antibiotics, hypertonic saline and β-adrenergic receptor agonists are used to control bacterial disease, increase hydration of airway surfaces, and improve lung function.25,26 Inhaled medications, such as albuterol, have been linked to decreased salivary pH and buffering capacity, and a subsequent increase in caries risk among individuals with other respiratory diseases, such as asthma.27,28 Many of these medications also induce xerostomia, further compounding caries risk. Oral antibiotics used to treat CF-related infections are often dosed in sugary suspensions to mask the bitter taste. The sugary suspension may coat the teeth for hours between toothbrushing and, thus, increase caries risk.29
Individuals with CF have extra health care needs30,31 that may leave families with few resources to obtain needed dental care. A recent study found that Medicaid-enrolled children with CF were significantly less likely to use dental care than Medicaid-enrolled children without CF.32 Frequent and unexpected hospitalizations are a barrier to scheduling dental appointments. Multidisciplinary CF teams often do not include dentists or dental hygienists. With so many competing demands on time and resources, dental care may end up becoming a low priority for families affected by CF. Inadequate or irregular use of dental care may increase caries risk.
CURRENT KNOWLEDGE OF CARIES
While these risk factors indicate that individuals with CF should be at high risk for caries, lower caries prevalence rates have been reported for CF populations compared to non-CF matched controls,7,20,33–36 healthy siblings,8,33 and individuals with chronic respiratory conditions.9 These findings gave rise to the notion that patients with CF are at low risk for caries. Possible protective factors include long-term antibiotic use, administration of pancreatic enzyme replacement therapy, and increased salivary buffering capacity.6,20,37–39
A 2013 systematic review reexamined the literature that supposedly supports the idea that individuals with CF are at reduced caries risk.40 A number of studies reported no difference in caries rates by CF status,9,41 and several demonstrated higher caries prevalence among individuals with CF.42–44 The review found that young children with CF appeared to have lower caries rates than other children, whereas protection against caries was lost during adolescence. New studies are underway to test this hypothesis — which has the potential to change the caries risk paradigm regarding individuals with CF.
CLINICAL CONSIDERATIONS
Preventive care for individuals with CF should center on specialized dietary counseling, oral hygiene instruction, and increased exposure to topical fluorides. Calorie-rich alternatives to fermentable carbohydrates include meats, nuts and cheeses, along with a balanced diet of fruits and vegetables. Eating frequency should also be considered, and may be accompanied by a recommendation to increase the frequency of toothbrushing with fluoride toothpaste. In addition, anticipatory guidance should be provided, as patients with CF are susceptible to calculus formation due to the elevated calcium and phosphorus content of their saliva caused by the CFTR mutation.45 This may necessitate more frequent hygiene appointments and close monitoring of the periodontal health of patients with CF. These patients should also be exposed regularly to topical fluorides to strengthen tooth enamel. Systemic fluoride intake should be monitored and proper dosing encouraged.
Since the pathophysiology of CF is most significant in the respiratory system, dental professionals should follow the same precautions used on other patients with compromised respiratory systems. Appointments should be kept short, and clinicians should allow for frequent breaks to ensure patient comfort. Positioning in the dental chair should be as upright as possible to aid in the clearing of secretions from the bronchi and trachea through frequent coughing.6 Nonsibling dental patients with CF should be scheduled at different times to help prevent patient-to-patient cross-infection with different strains of the lung-damaging bacteria Pseudomonas aeruginosa.46
Dental professionals should be aware of the challenges in sedating individuals with CF. Children with CF are poor candidates for in-office oral conscious sedation because of compromised lung function. The use of agents that interfere with pulmonary function, such as sedatives and narcotic analgesics, is contraindicated.6 Historically, treatment under general anesthesia was contraindicated in this population because of adverse effects reported after surgery. Modern anesthetic techniques using new compounds, such as isoflurene and sevoflurane, however, do not adversely affect lung function in children with mild to moderate CF.47 Oral rehabilitation under general anesthesia should be performed in a hospital setting, and only undertaken after thorough consultation with a patient’s primary care physician and CF management team. Likewise, nitrous oxide inhalation was historically contraindicated in patients with CF, but the medical community has rediscovered it as a helpful tool for managing anxiety and pain.48 For individuals with CF whose lungs have not progressed to obstructive airway disease or emphysema, nitrous oxide may be used to manage anxiety and limit the need for other sedation methods — such as oral, intramuscular or intravenous conscious sedation or general anesthesia.
CONCLUSION
Advances in early diagnosis and treatment have dramatically increased the survival of children and adolescents with CF. Dental professionals should be cognizant of the unique characteristics this genetic condition poses for caries risk. Prevention is paramount because oral disease may have detrimental effects in immunocompromised patients. Dental teams should stress aggressive preventive measures and recognize special treatment considerations in order to support the overall health of patients with CF.
References
- Lipuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23:299–323.
- Strausbaugh SD, Davis PB. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med. 2007;28:279–288.
- Cystic Fibrosis Foundation. Patient Registry Annual Data Report 2012. Available at: cystic fibrosisdata.org/ReportsUS.html. Accessed July 7, 2016.
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001.
- Accurso FJ, Sontag MK, Wagener JS. Complications associated with symptomatic diagnosis in infants with cystic fibrosis. J Pediatr. 2005;147(3 Suppl):S37–S41.
- Fernald GW, Roberts MW, Boat TF. Cystic fibrosis: a current review. Pediatr Dent. 1990;12:72–78.
- Primosch RE. Tetracycline discoloration, enamel defects, and dental caries in patients with cystic fibrosis. Oral Surg Oral Med Oral Pathol. 1980;50:301–308.
- Jagels AE, Sweeney EA. Oral health of patients with cystic fibrosis and their siblings. J Dent Res. 1976;55:991–996.
- Narang A, Maguire A, Nunn J, Bush A. Oral health and related factors in cystic fibrosis and other chronic respiratory disorders. Arch Dis Child. 2003;88:702–707.
- Azevedo T, Feijo G, Bezerra A. Presence of developmental defects of enamel in cystic fibrosis patients. J Dent Child (Chic). 2006;73:159–163.
- Ferrazzano G, Sangianantoni G. Dental enamel defects in Italian children with cystic fibrosis: an observational study. Community Dent Health. 2012;29:106–109.
- Arquitt CK, Boyd C, Wright JT. Cystic fibrosis transmembrane regulator gene (cftr) is associated with abnormal enamel formation. J Dent Res. 2002;81:492–496.
- Bronckers A, Kalogeraki L, Jorna HJN, et al. The cystic fibrosis transmembrane conductance regulator (CFTR) is expressed in maturation stage ameloblasts, odontoblasts and bone cells. Bone. 2010;46:1188–1196.
- Mousa HM, Woodley FW. Gastroesophageal reflux in cystic fibrosis: current understandings of mechanisms and management. Curr Gastroenterol Rep. 2012;14:226–235.
- Vic P, Tassin E, Turck D, Gottrand F, Launay V, Farriaux JP. Frequency of gastroesophageal reflux in infants and in young children with cystic fibrosis. Arch Pediatr. 1995;2:742–746.
- Blondeau K, Pauwels A, Dupont L, et al. Characteristics of gastroesophageal reflux and potential risk of gastric content aspiration in children with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2010;50:161–166.
- Alfaro EV, Aps JKM, Martens LC. Oral impli ca – tions in children with gastroesophageal reflux disease. Curr Opin Pediatr. 2008;20:576–583.
- Ersin NK, Gülen F, Eronat N, et al. Oral and dental manifestations of young asthmatics related to medication, severity and duration of condition. Pediatr Int. 2006;48:549–554.
- Linnett V, Seow WK, Connor F, Shepherd R. Oral health of children with gastro-esophageal reflux disease: a controlled study. Aust Dent J. 2002;47:156–162.
- Kinirons MJ. Increased salivary buffering in association with a low caries experience in children suffering from cystic fibrosis. J Dent Res. 1983;62:815–817.
- Catalán MA, Scott-Anne K, Klein MI, Koo H, Bowen WH, Melvin JE. Elevated incidence of dental caries in a mouse model of cystic fibrosis. PLoS One. 2011;6:e16549.
- Smyth AR, Bell SC, Bojcin S, et al. European Cystic Fibrosis Society standards of care: best practice guidelines. J Cyst Fibros. 2014;13S1:S23–S42.
- Woestenenk JW, Castelijns SJ, van der Ent CK, Houwen RHJ. Nutritional intervention in patients with cystic fibrosis: a systematic review. J Cyst Fibros. 2012;12:102–115.
- Moursi AM, Fernandez JB, Daronch M, Zee L, Jones CL. Nutrition and oral health considerations in children with special health care needs: implications for oral health care providers. Pediatr Dent. 2010;32:333–342.
- Flume PA, O’Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957–969.
- Halfhide C, Evans HJ, Couriel J. Inhaled bronchodilators for cystic fibrosis. Cochrane Database Syst Rev. 2005;19:CD003428.
- Milano M, Lee JY, Donovan K, Chen JW. A cross-sectional study of medication-related factors and caries experience in asthmatic children. Pediatr Dent. 2006;28:415–419.
- Kargul B, Tanboga I, Ergeneli S, Karakoc F, Dagli E. Inhaler medicament effects on saliva and plaque pH in asthmatic children. J Clin Pediatr Dent. 1998;22:137–140.
- Donaldson M, Goodchild JH, Epstein JB. Sugar content, cariogenicity, and dental concerns with commonly used medications. J Am Dent Assoc. 2015;146:129–133.
- Berlinski A, Chambers MJ, Willis L, Homa K, Com G. Redesigning care to meet national recommendation of four or more yearly clinic visits in patients with cystic fibrosis. BMJ Qual Saf. 2014;23(Suppl 1):i42–i49.
- Ditto MR, Jones JE, Sanders B, Weddell JA, Jackson R, Tomlin A. Pediatrician’s role in children’s oral health: an Indiana survey. Clin Pediatr (Phila). 2010;49:12–19.
- Sarvas EW, Huebner CE, Scott JM, Aps JKM, Chi DL. Dental utilization of Medicaid-enrolled children with cystic fibrosis. J Dent Res. 2015;94(Sp Iss A):4202.
- Aps JK, Martens LC. [Oral health risks in patients with cystic fibrosis]. Rev Belge Med Dent (1984). 2004;59:114–120.
- Kinirons MJ. The effect of antibiotic therapy on the oral health of cystic fibrosis children. Int J Paediatr Dent. 1992;2:139–143.
- Kinirons MJ. Dental health of patients suffering from cystic fibrosis in Northern Ireland. Community Dent Health. 1989;6:113–120.
- Ferrazzano GF, Orlando S, Sangianantoni G, Cantile T, Ingenito A. Dental and periodontal health status in children affected by cystic fibrosis in a southern Italian region. Eur J Paediatr Dent. 2009;10:65–68.
- Atar M, Körperich EJ. Systemic disorders and their influence on the development of dental hard tissues: a literature review. J Dent. 2010;38:296–306.
- Sweeney EA, Shaw JH. The effect of dietary pancreatin supplements on dental caries and on the composition of saliva in caries-susceptible rats. J Dent Res. 1965;44:973–976.
- Littleton NW, White CL. Dental findings from a preliminary study of children receiving extended antibiotic therapy. J Am Dent Assoc. 1964;68:520–525.
- Chi DL. Dental caries prevalence in children and adolescents with cystic fibrosis: a qualitative systematic review and recommendations for future research. Int J Paediatr Dent. 2013;23:376–386.
- Aps JK, Van Maele GO, Claeys G, Martens LC. Mutans streptococci, lactobacilli and caries experience in cystic fibrosis homozygotes, heterozygotes and healthy controls. Caries Res. 2001;35:407–411.
- Storhaug K. Caries experience in disabled pre-school children. Acta Odontol Scand. 1985;43:241–248.
- Storhaug K, Holst D. Caries experience of disabled school-age children. Community Dent Oral Epidemiol. 1987;15:144–149.
- Dabrowska E, Błahuszewska K, Minarowska A, Kaczmarski M, Niedźwiecka-Andrzejewicz I, Stokowska W. Assessment of dental status and oral hygiene in the study population of cystic fibrosis patients in the Podlasie province. Adv Med Sci. 2006;51(Suppl 1):100–103.
- Wotman S, Mercadante J, Mandel ID, Goldman RS, Denning C. The occurrence of calculus in normal children, children with cystic fibrosis, and children with asthma. J Periodontol. 1973;44:278–280.
- Kidd TJ, Soares Magalhães RJ, Paynter S, Bell SC. The social network in cystic fibrosis centre care and the risk of shared Pseudomonas aeruginosa strain infection: a cross-sectional analysis. Lancet Respir Med. 2015;3:640–650.
- Pandit C, Valentin R, De Lima J, et al. Effect of general anesthesia on pulmonary function and clinical status on children with cystic fibrosis. Paediatr Anaesth. 2014;24:164–169.
- Williams V, Riley A, Rayner R, Richardson K. Inhaled nitrous oxide during painful procedures: a satisfaction survey. Paediatr Nurs. 2006;18:31–33.
From Decisions in Dentistry. August 2016;2(08):34–36,39.

